LLMs and Antibodies
Published:
motivation
I wanted to understand if LLM / PLMs can be useful for analyzing antibody sequence data. Antibodies are proteins made by the immune system which are critical for protection against viruses. Here I used antibody sequencing data, AbLang (Antibody LLM), and single-cell rna sequencing data investigate.
protein language models
Protein language models are inspired by natural language processing (NLP) models like GPT-4. They are designed to analyze and predict protein sequences, which are chains of amino acids with distinct functions in living organisms. These models are trained on large datasets of known protein sequences and appear to learn general truths and patterns about protein space through relatively simple and unsupervised training procedures. By understanding these patterns, the models can predict protein structure and function. Similar to NLP models, protein language models use a technique called “attention” to weigh the importance of different amino acids in the sequence, allowing them to capture long-range interactions and dependencies between amino acids. I wanted to understand how these models can be applied to studying antibody evolution in particular.
Ablang Description
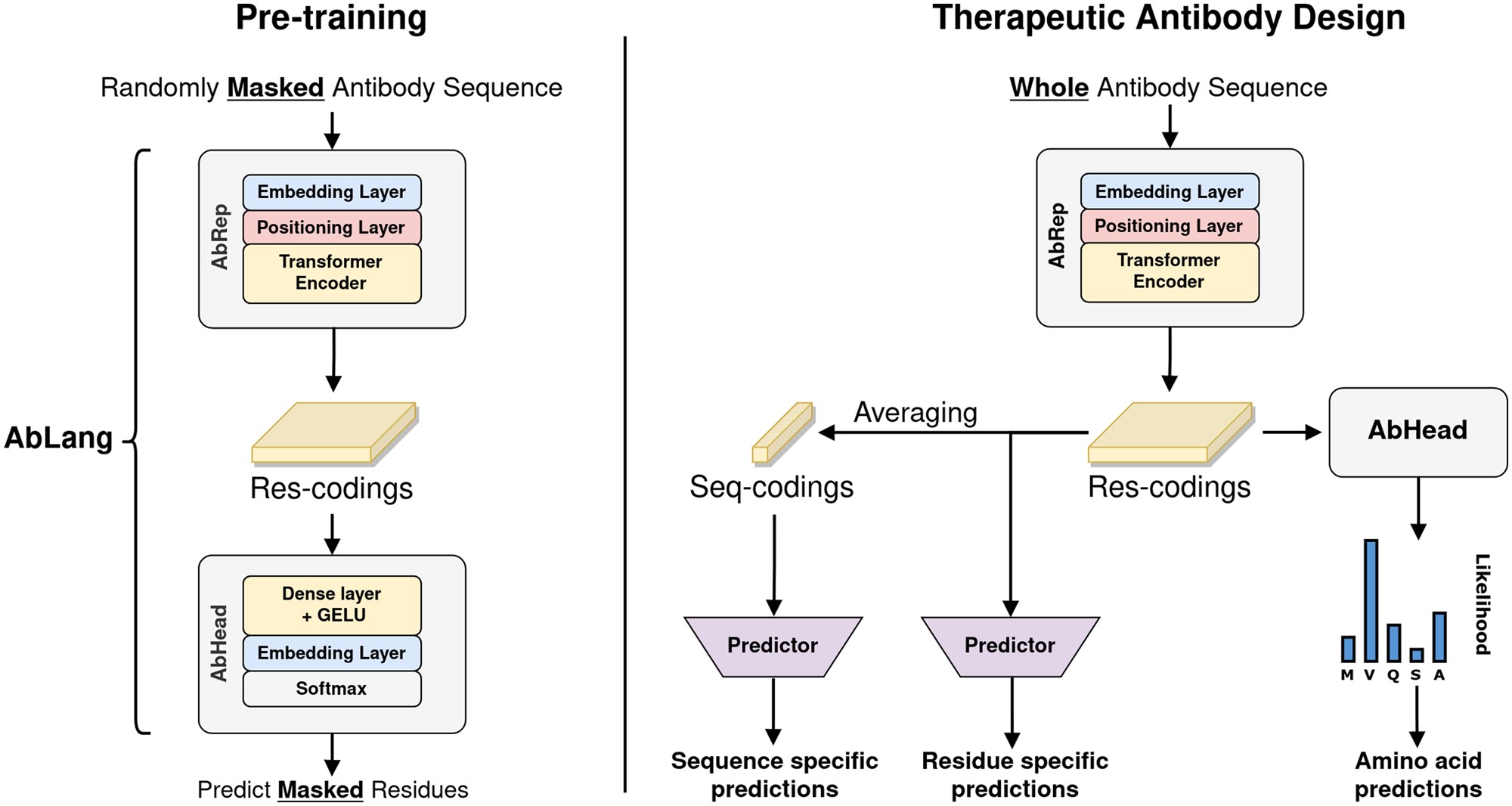
Ablang is a protein language model trained specifically on antibodies which was readily available to me and easy to install at the time. There’s a strong argument to made for using a more general protein language model. At the least, one should compare the results here to something like ESM from Meta.
Additional Background about Antibodies at the bottom:
All that you’d need to know to understand what I’m going to show in this notebook is that generally speaking more somatic hypermutation means the antibody in question has been through more of this evolutionary process than an antibody that has no somatic hypermutation
Overview of my investigation:
- associate the single cell transcriptome data with the embeddings calculated using AbLang for each B cell
- perform PCA on the LLM embeddings
- ad-hoc generation separation of similar antibody-types in the PCA space
- differential gene-expression analysis of B cells using these LLM defined clusters
- visualize embeddings with UMAP
LLMs, Antibodies, and Single Cell Genomics
A Quick Look at the Dataset
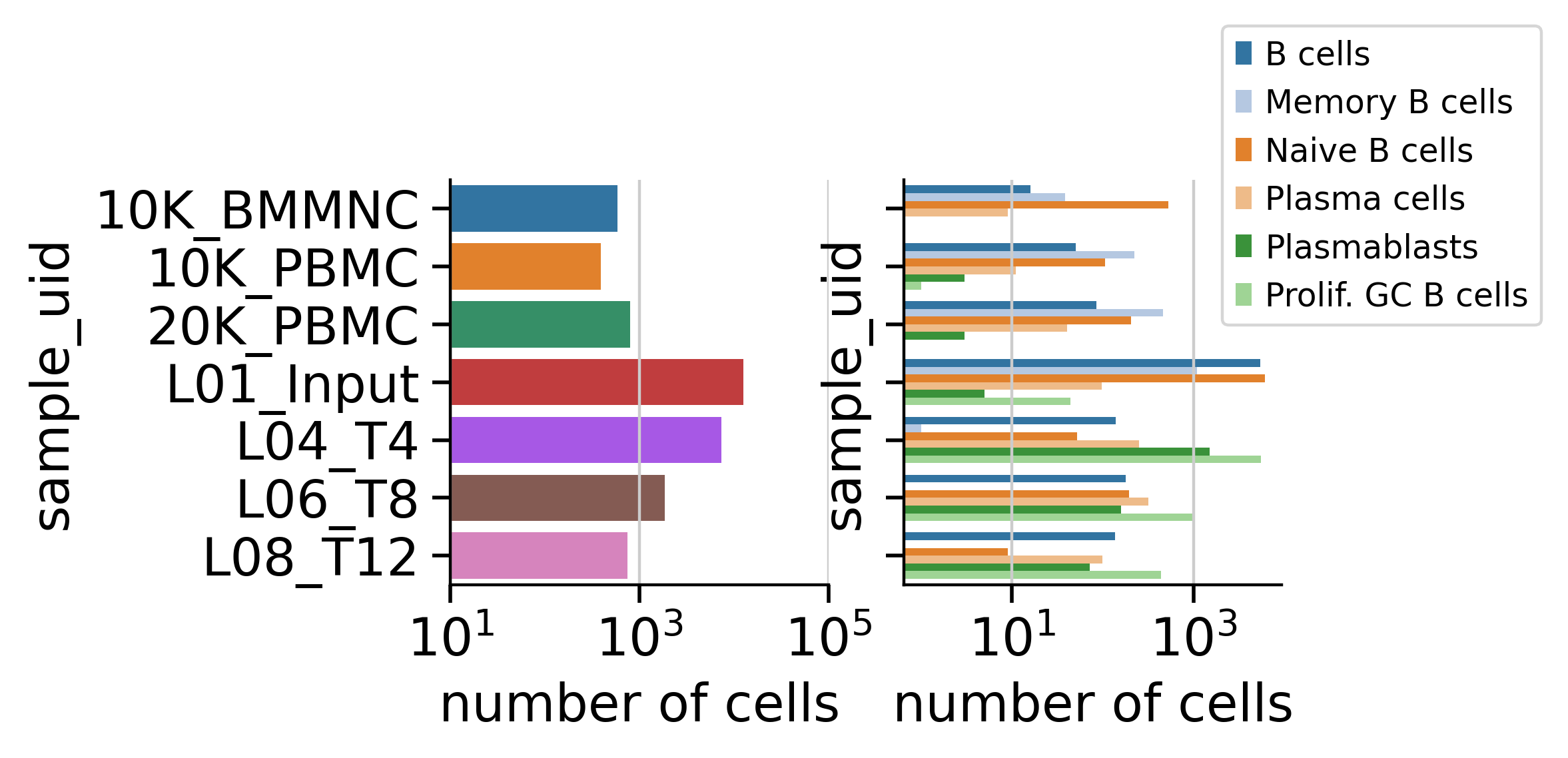
So we have hundreds to thousands of single B cells in each sample with each B cells having an RNA transcriptome and an Antibody Sequence associated with it
I’m hoping this helps orient – we can see the single cell RNA sequencing data coheres in a global way with the Antibody Sequence via these UMAPs:
- I’m plotting UMAP projections of the single cell RNA sequencing data colored by metadata about the transcriptional state (far left) or the antibody sequence (right two)2. Memory B cells have previously participated in immune respones and thus have acquired hypermutations in the antibody sequence. Naive B cells have no mutation in their antibody sequence.
- The antibody mutation level is determined by asking how far away any given antibody is in nucleotide spacefrom our best guess of the VDJ genes which constructed it.
- Memory B cells also appear to have switched Antibody Constant Region usage, another sign of they have already participated in the immune response.
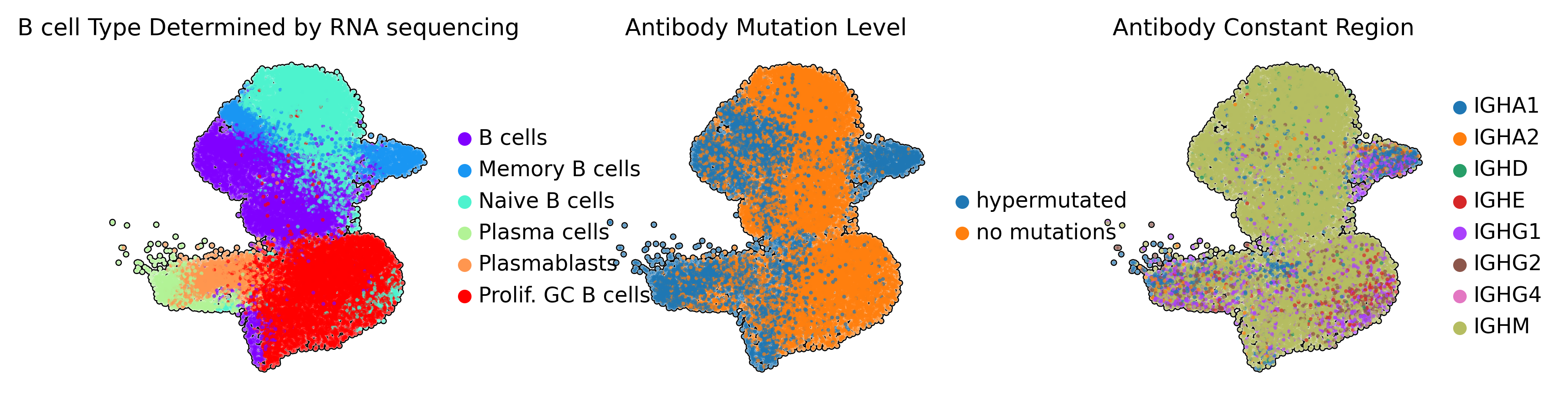
We can see my annotations of the antibody sequence are clearly related in a global way to the B cell Type determined via single-cell RNA sequencing. For example, cells which are Memory B cells most ofter have antibody sequences I annotated as hypermutated.
I’m curious whether the sequence encoding of the antibody sequence can also meaningfully represent a known phenomenon like hypermutation or constant region usage.Now I’m plotting the same categorical variables from the UMAPS on the PCA of the antibody sequence embedding calculated using AbLang
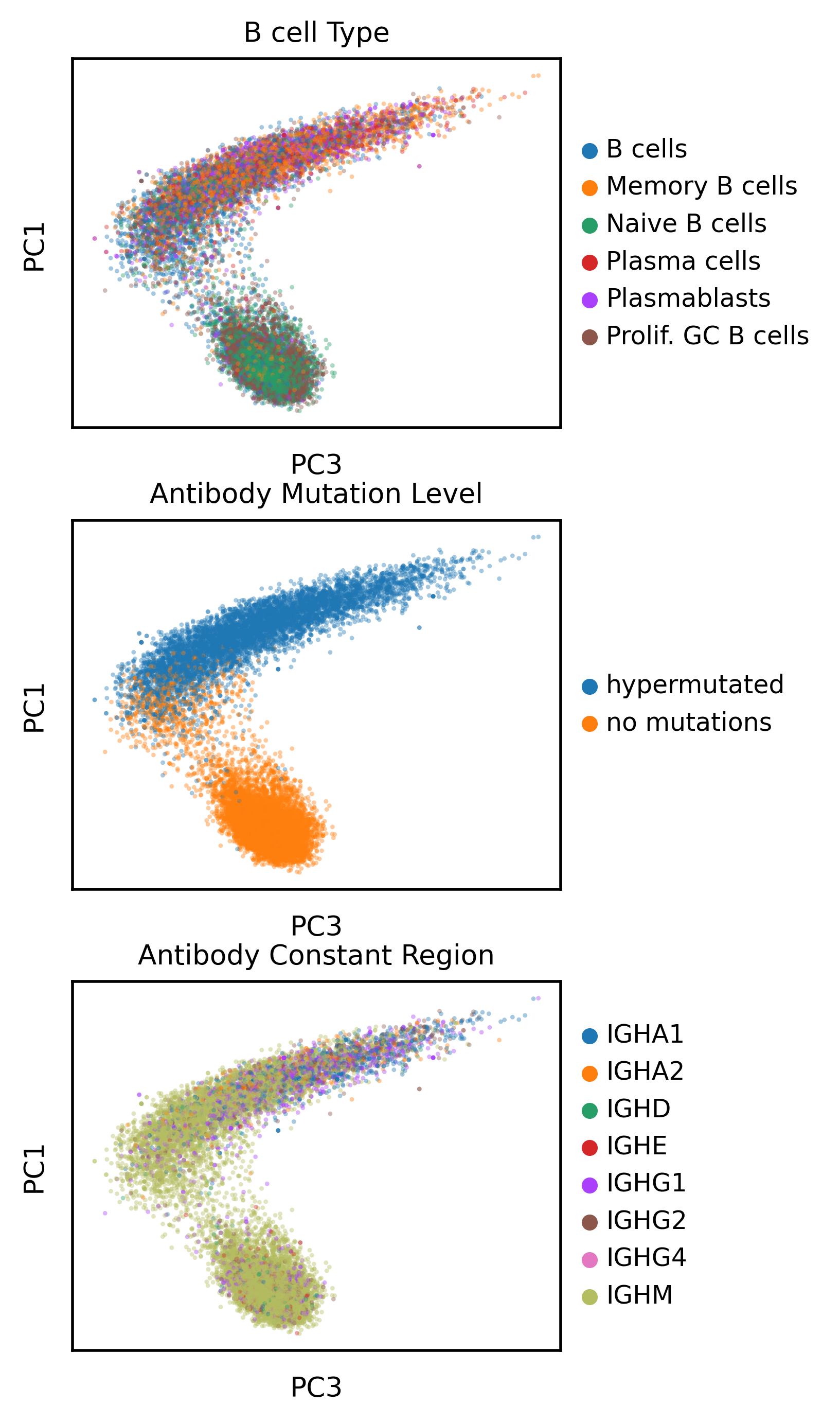
This is interesting, it is clear the language model has learned what hypermutation looks like and pretty neatly separates antibodies with no mutations from mutated antibodies using PC1 and PC3. Furthermore, on PC3, there appears to be continuous varation in the hypermutated antibody space, but it’s clear there is an arrow that can be drawn from predominantly IGHM class antibodies to the other type (e.g. IGHA1). So the model appears to capture something meaningful about antibodies.
Let’s look at the relationship between hypermutation and the the PCs in a quantitative sense, in particular I’m interested in PC3, which appears to separate IGHM antibodies from the others. Does this separation exist in a meaningful way in gene expression space as well?
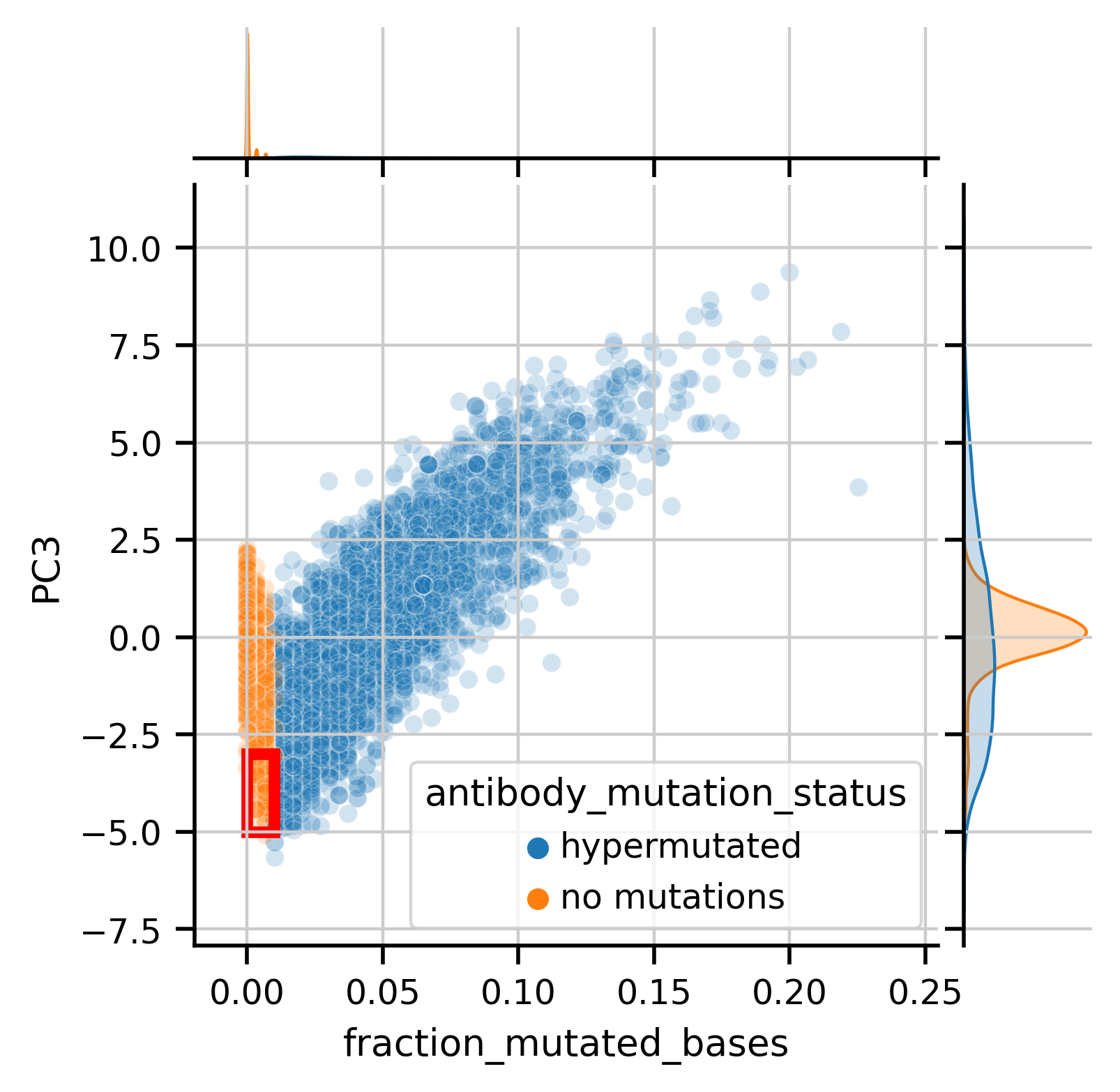
As an aside, we can see a fair amount of “no mutation” antibody sequences (highighted in the red box or seen in the tail of the orange distribtion) that actually appear more like mutated antibodies in the PC space. I asked if the gene expression of cells with that profile looked like Memory B cells, which would corroborate that the antibody actually is hypermutated. Indeed these cells with “no mutations” appear to have a memory-like phenotype, suggesting the language model identifies mutations in antibodies more sensitively than how I orginally labeled the mutation status, via comparison to a genomic database.
I noticed from some exploratory plots that the while the distribution of hypermutated antibodies on PC3 doesn’t appear to have any notable features, if I subset different cell types, multiple modes appear. For example, hypermutated Memory B cells appear to have multple two modes in PC3
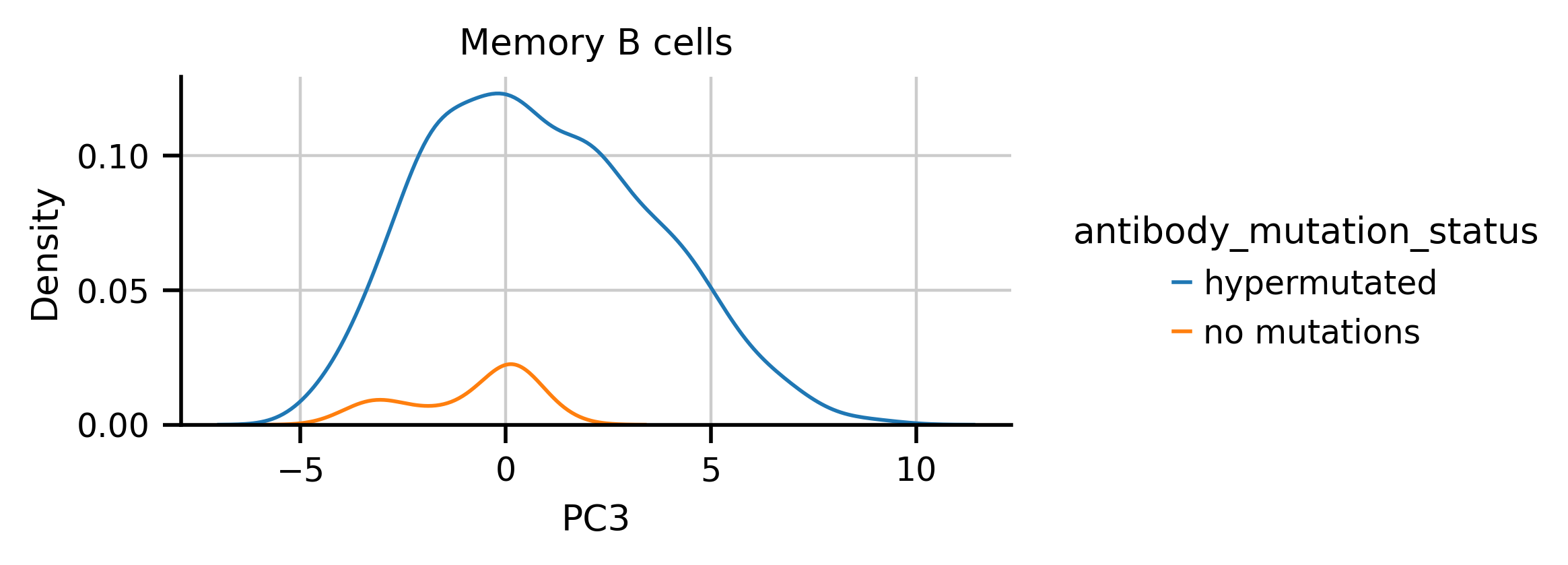
I split the hypermutated Memory B cells at 1 in PC3 space and asked if there were gene expression differences between the groups
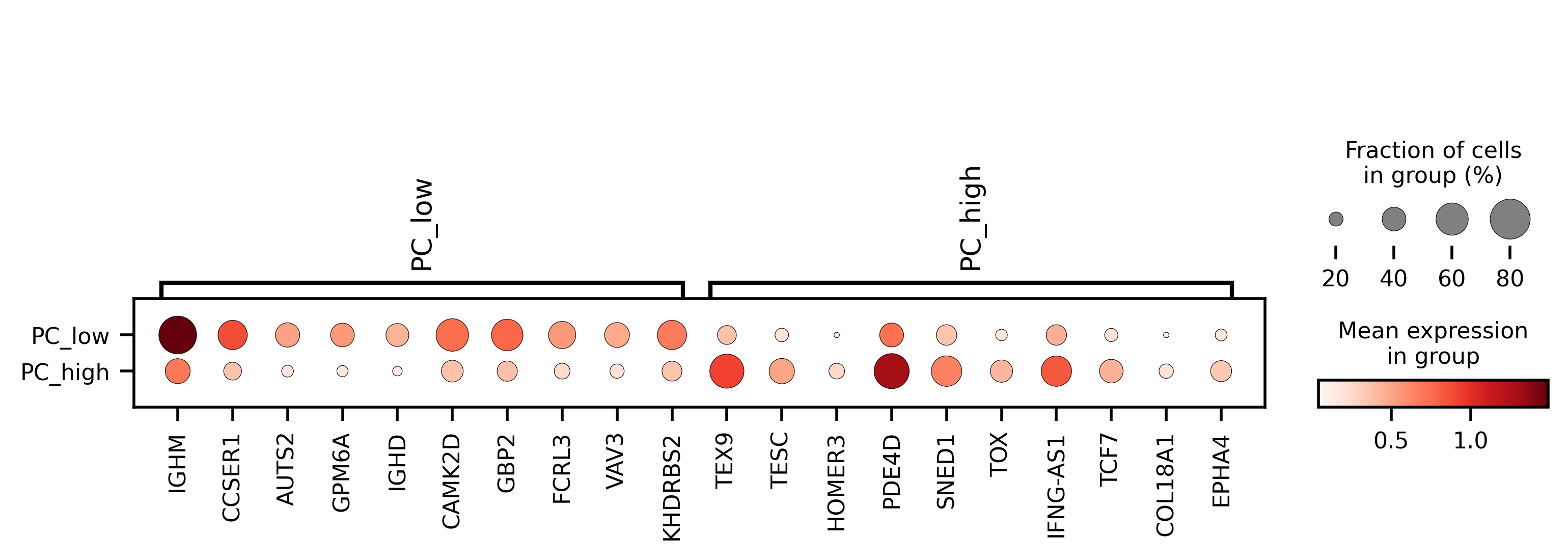
Indeed there are clear differences, with PC3 low having a IGHM memory phenotype and PC3 high deeper or class-switched memory phenotype
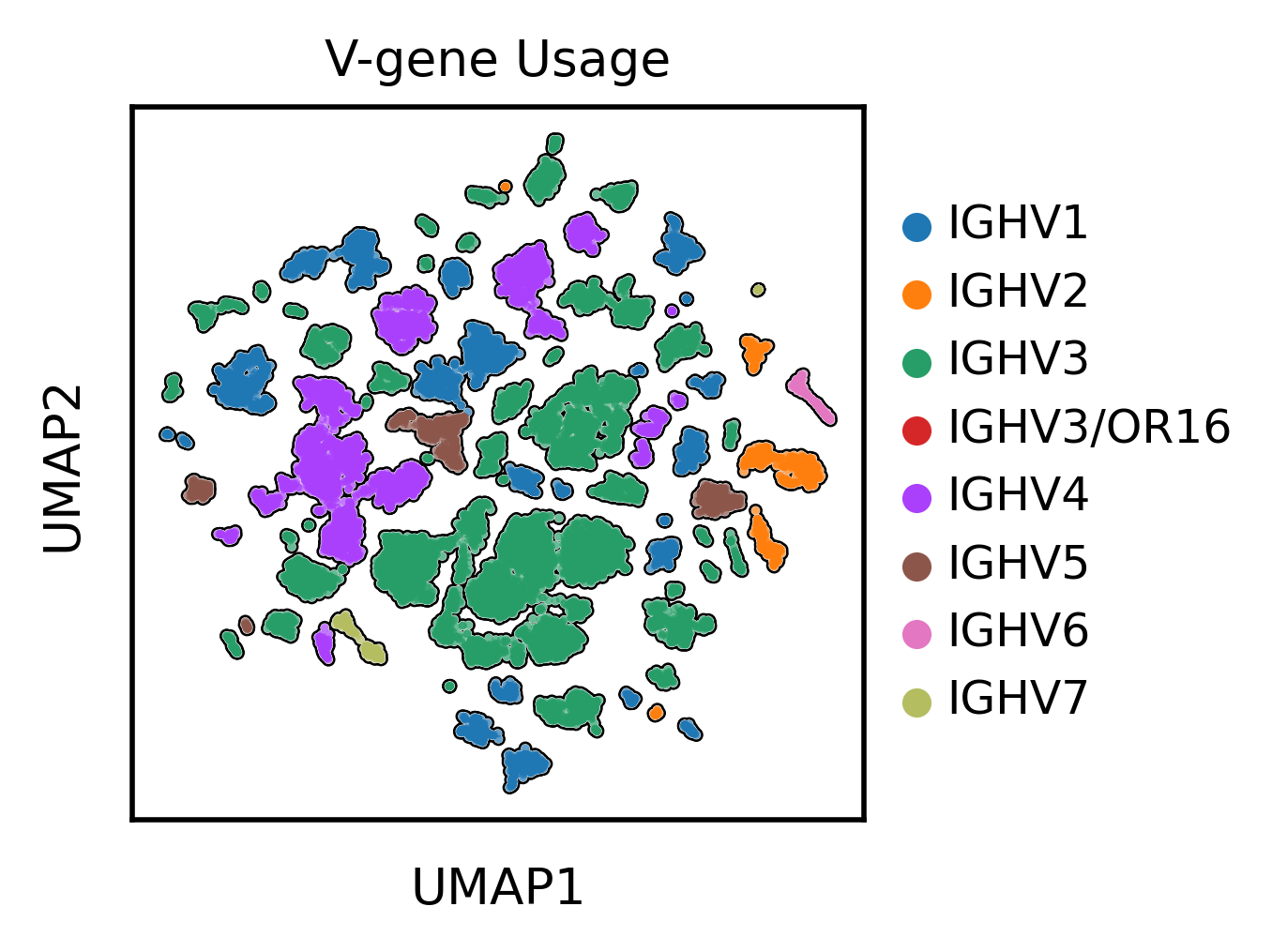
Let’s finally take a quick look at the UMAP projection of the antibody sequence encodings
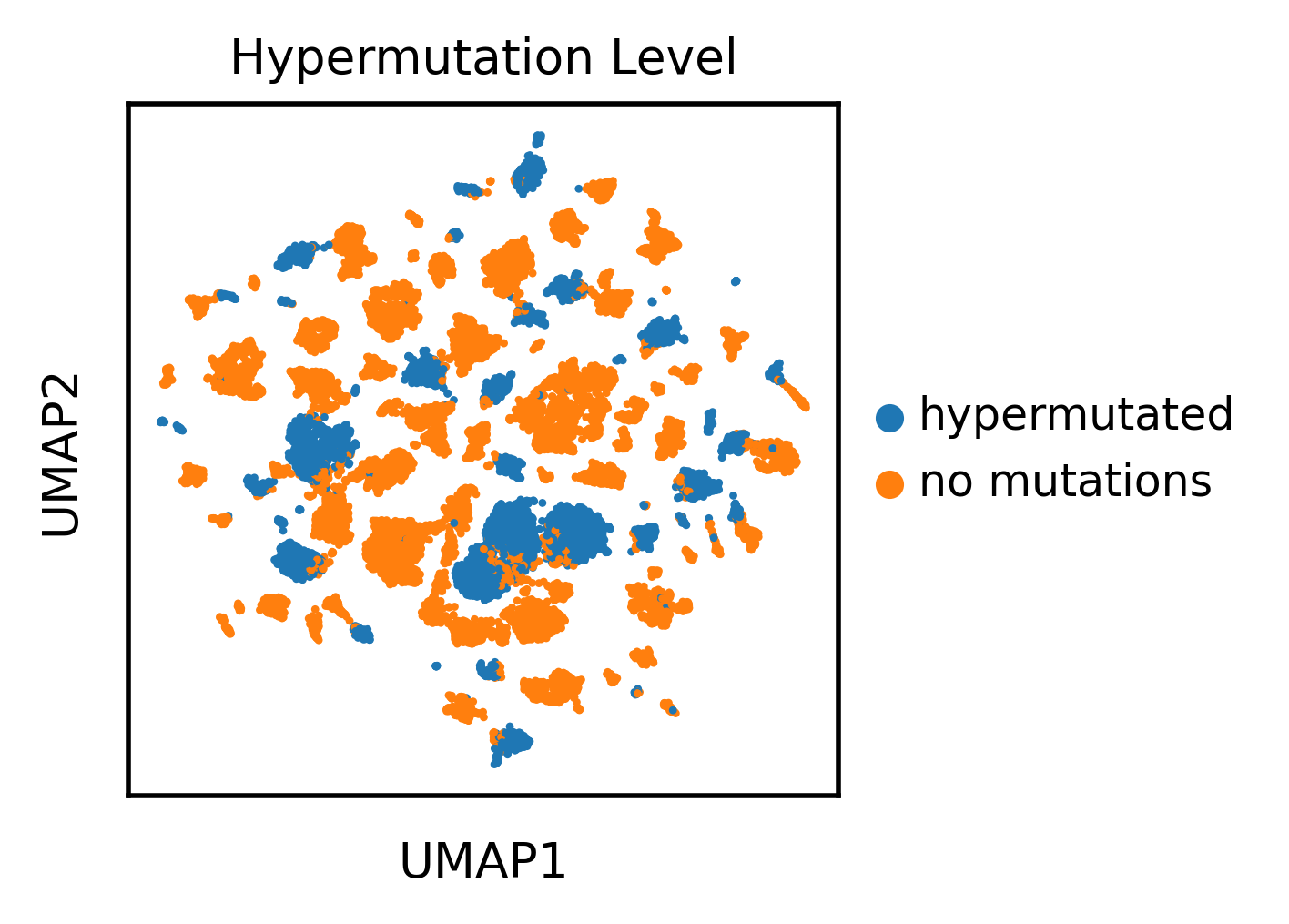
Looks like the UMAP separates antibody sequences by V gene usage in addition to the hypermutation levels.
We can see that the same V gene generally clusters together, but within these broad gene families the mutated sequences cluster together or apart from the no mutations sequences
Summary:
I was curious about large language models, so I downloaded one and applied it to data I’ve generated, I found that:
- the language model encodes antibody hypermutation on 2 relatively orthongonal axes
- one is just whether or not there are any mutations
- the other is how many mutations there are
- ad-hoc classification of antibody encoding types generally coheres with my orthogonal measurement of gene expression
To do:
- Investigate the distance in antibody encoding space between related antibody sequences, is that distnace meaningfully smaller than a reasonable null expection?
- train LLM on all of antibody space (e.g. mouse, camelid, monkey) vs just human (which Ablang has done). It’s a poorly formed idea, but this could be used to perform comparative immunogenomics in a less-biased manner
- train LLM only on memory antibody space (Ablang has used unmutated and mutated antibodies and I wonder if the large amount of unmutated antibodies may obscure something about hypermuation and antibody evolution)
Antibody Background
An antibody is a protective protein produced by the B cells in the immune system to neutralize and defend against harmful pathogens, such as viruses. Structurally, antibodies are composed of four polypeptide chains—two identical heavy chains and two identical light chains—linked by disulfide bonds, forming a Y-shaped m/images/posts/nest-map/olecule. The unique feature of antibodies lies in their variable region, located at the tips of the Y-shaped structure, which is responsible for binding specifically to the foreign substance, or antigen.
The variable region is generated stochastically through a process called V(D)J recombination. This process involves the pseudo-random selection, recombination, and joining of gene segments—Variable (V), Diversity (D), and Joining (J) segments—along with the introduction of pseudo-random nucleotide insertions and deletions between the recombined genes. This generates astromomical standing antibody sequence diversity which could never be encoded in the genome. Selection of particular antibodies from this diversity allows indivuduals to adapt their immune system to never-before-seen pathogens.
Antibody Evolution
The antibody generation process creates a large pool standing diversity in protein space. Next, the immune system uses an evolutionary process to select and amplify the antibodies which have the most beneficial properties, such as strongly binding and neutralizing a pathogen which is wreaking havoc in your body. During the selection process, additional diversity is generated by mutating the selected antibodies, allowing the immune system iterate on creating an even better protein: it selects the best of the selected. This process is called somatic hypermuation, and it’s pretty much fascinating.