RNA Obelisks
Published:
novel viroid-like entities revealed by computational biology
These authors would write this paper, and I mean that in the good way. We have Andy Fire, who is famous for a deeply thoughtful career in RNA biology. We have Ami Bhatt: a world-class researcher who leads a group that generates and interprets micro-biome sequencing data. We also have the participation of Robert C. Edgar on the paper — he is, as far as I am aware, the only “independent researcher” in biology that is respected. His open-source software MUSCLES its way into many a phylogenetic analysis. Plus I like his style:
 this is the platonic case of a dude who got rich by selling a tech company in the late 90s
this is the platonic case of a dude who got rich by selling a tech company in the late 90s
So what’s a viroid?
It is like a virus, but even simpler. Viruses are genetic parasites which generally carry nothing more than what is required to self-replicate. This includes their own polymerase which copies their genome and a protein coat which envelopes the genome. Viral polymerases are often used to identify and classify viruses via sequence similarity. Of course, if you’re a freeloading parasite already, why not freeload harder and just use your host’s polymerase and also it’s warm inside cells so get rid of that protein coat. To me, seems like a winning strategy. Yet this strategy would make traditional approaches for detecting viroid-like entities difficult, because they don’t have much sequence similarity to anything else. And they don’t have much sequence at all really.
Viroid nominator
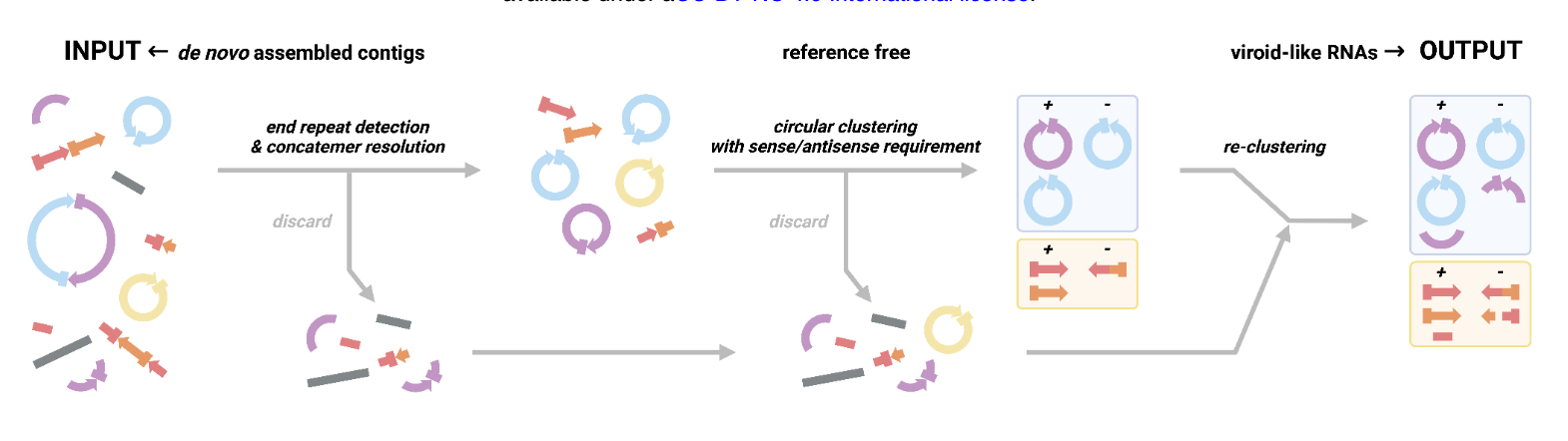
Enter Vnom a program developed by the authors which uses a set of heuristics to nominate possible viroid-like elements. Possible candidate assemblies should be circular and detected in positive and negative strand orientation (indicative of replication)
The authors note:
While these filters should enrich for viroid-like RNAs, highly repetitive sequences also satisfy these requirements and so are often also enriched. VNom was found to work adequately well on deeply sequenced viroid-positive plant RNA-seq datasets (e.g. SRR11060618, SRR11060619, SRR11060620, and SRR16133646), especially when assemblies from the same bioProject were grouped together.
As a reader, I would be curious to understand more about the outputs of this algorithm. Is there any way to estimate the specificity or sensitivity? Obviously the ground-truth isn’t known, but even downsampling the plant RNA-seq datasets mentioned would be interesting. Additionally, even a heavily caveated analysis of the outputs would be welcome. How many outputs appear to be highly repetitive, how many have ORFs, what is the homology of these ORFs to other proteins?
Obelisk-like RNA structure and donor variability:
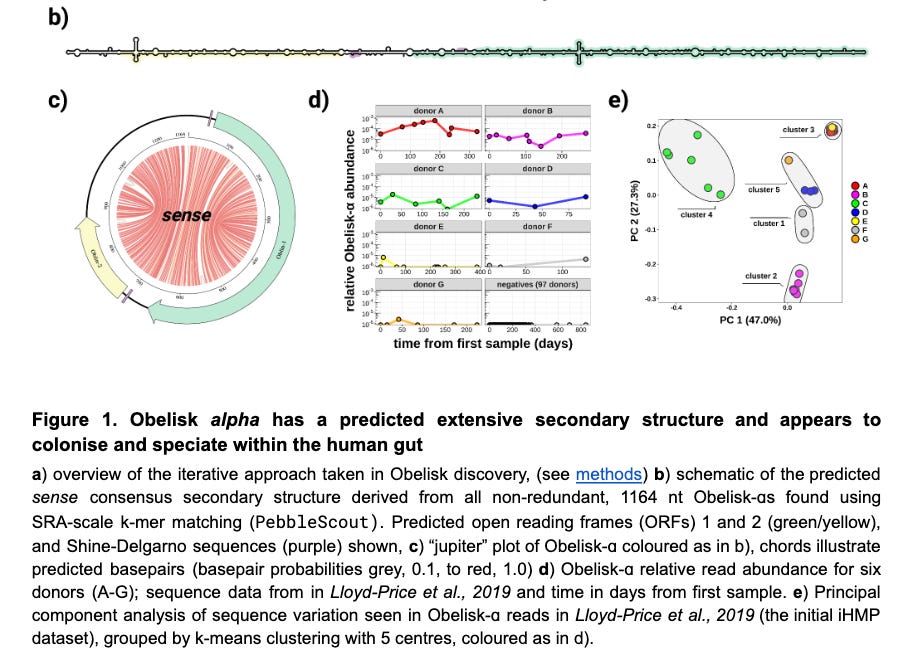
The authors predict the RNA secondary structure of these entities and find them to be highly “rod-like” or “obelisk-y” as seen in b and c. Figure c is a chord plot where the red lines denote predicted based pairing. I’ve never really found these plots useful in other areas of genomics, but this one does give a pretty good sense of the RNA structure. Figure d below is interesting as it shows within donor variability in Obelisk RNA read counts. The temporal stability of these entities within donors is clear. The abundance of in donor A is shocking to me — at ~190 days around 1/5000 reads belong to map to Obelisks.
What does the RNA make?
On the protein side, the authors make structural predictions of the Oblin-1 coding regions using AlphaFold. The globular folds for many of the representatives (Figure 4 tertiary structures) hint at potential RNA binding or chaperone functions (worth validating experimentally). Certainly experimental work is necessary, but the high degree of structural conservation despite sequence divergence points to an real function for these proteins. We can only speculate, but perhaps they are kinetically helpful for co-opting host transcriptional machinery.
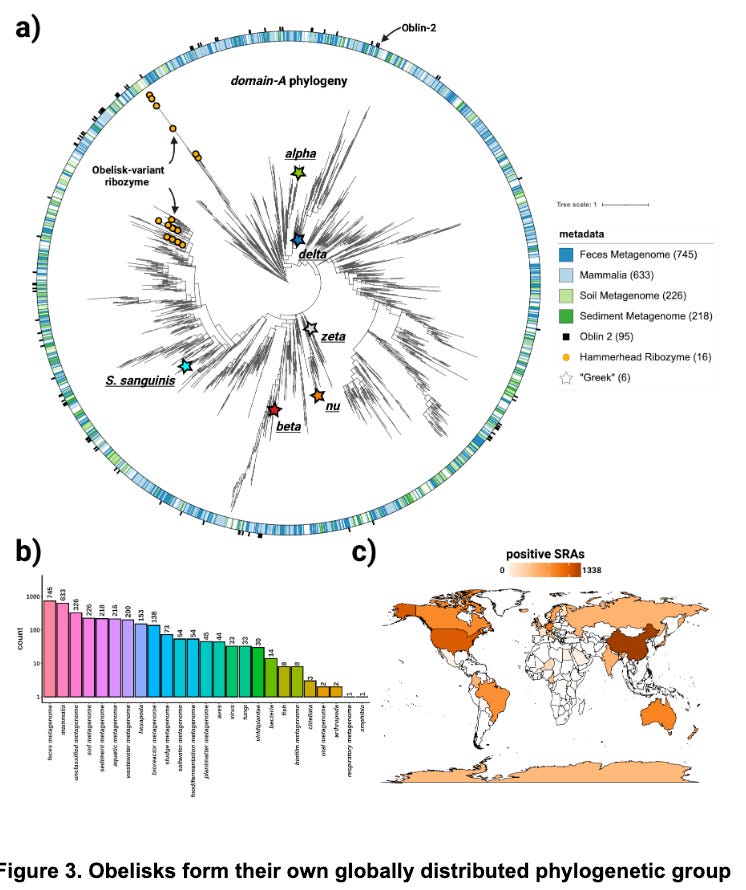
The proposed phylogeny (Figure 3a) is messy, and Robert would be the first to tell you it can be quite tricky to estimate correct phylogenetic topologies. The authors’ suggest new phylogenetic approaches that integrate RNA structure and protein folding with MSA could prove powerful not just for Obelisks but other structured RNAs as well. It is an interesting idea, people often incorporate some priors about models of what mutations are likely to occur, but I’ve only really seen it in the context of a biochemical model of how mutations occur. Regardless of the topology of the phylogeny, it is clear these RNA entities are all over the globe, which is cool.
Are they CRISPy?
The authors checked if any Obelisks have been captured by CRISPR systems, which may indicate hostile interactions with prokaryotic hosts. Using a database of nearly 30 million CRISPR spacers, they ran a conservative k-mer matching search. Out of around 39,000 Obelisk sequences examined, only one measly spacer mapped to a putative “Obelisk-gamma” contig over 1000 nts long. Seems this spacer comes from the alphaprotebacterium Bombella mellum. But Obelisk-gamma looks kinda wonky - deviating from that classic rod-like structure with some unpaired, frayed ends. The author’s remain equivocal about whether this is some type of computational artefact or a real CRISPR hit.
Given caveats about the sensitivity of sequence similarity search, I’m not ruling out that CRISPR system occasionally snag an Obelisk. And I’m not sure we’d be surprised not to find them anyways. There’s some math about their prevalence plus how many spacers are in the database and whether the database is biased in some way. However, their absence raises an interesting question of whether these RNAs have a special ability able to evade the prokaryotic immune system effectively .. or maybe they aren’t deletrious enough to merit mounting an immune response against.
Discussion
Overall, this work reveals a novel class of putative sub-viral entities that appear to be prevalent in the human microbiome. The Obelisks’ distinct structures, coding properties, and inferred evolutionary dynamics set them apart from classical viroids and other sub-viral entities. Mostly questions remain. What is the nature of their interactions with their hosts? What do Oblins do and what might this have to do with replication strategies? Are they able to actively avoid CRISPR systems or was this a limitation in sampling / sequence analysis sensitvity? Could there be useful and unique molecular functionality discovered by biochemically characterizing these or other sub-viral entities?
Regardless of whether there is wet-lab follow-up, this is precisely the kind of discovery that gets me excited about the era of big genomics data and the power of creative computational bio-prospecting. Here instead of reporting an incrementally better single-cell clustering algorithm, the authors instead reveal new branches of the tree of life.